Week 9: More Validation (Cont.)
May 2, 2025
This week, I ran another (likely my final) western blot. Because I have more material from the very first immunoprecipitation I did (the one I labeled “i,” see Week 3’s blog), I checked the first gel I ran for mass spectrometry (which failed because it was uneven) and used the 3rd replicate of each construct as it was the most consistent.
I ran my western with SM (without reducing reagent) and i, using the 3rd replicate of each. This time, instead of verifying for TMEM106B first, I focused on cathepsin D (sc-13148, mouse 1:500) and cathepsin A (Proteintech 15020-1-AP, rabbit 1:1000).
Because I had to leave early, I asked another member of the lab to help complete the western blot transfer onto a membrane, to which he graciously agreed. He left my membrane in EveryBlot blocking buffer in the cold room overnight.
The next day, I took my membrane from the cold room and created/added my cathepsin D solution, then left it on the shaker for 1 hour. After washing with TBST for 30 minutes, I added my cathepsin A solution onto the membrane and left it on the shaker for another hour, before washing with TBST for another 30 minutes.
I added my secondary antibody solution made of 2µL mouse and rabbit antibody each and EveryBlot blocking buffer and left it on the shaker. Finally, I repeated the remaining steps of a western (washing).
When I went to image my membrane, the bands were so faint that I had to dramatically turn up the intensity to see them, until the background noise was huge. I separated the scan into three separate images:
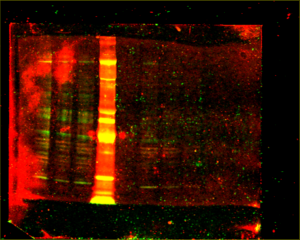
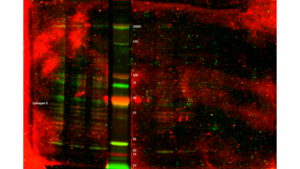
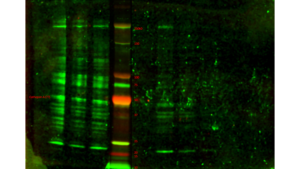
Before leaving for the day, I left the membrane in the cold room.
As there is way too much background noise, not enough signal, streaks in some lanes, and lots of non-specific binding (indicated by the presence of multiple bands in each lane), I had to change something about the way the antibodies bind to the membrane.
The next day, I stripped the membrane of its antibodies using stripping buffer. To do so, I washed the membrane with TBST, added the stripping buffer, incubated for 30min, washed with TBST again, then blocked with 5% milk in TBST for an hour. This allows me to start the antibody binding process from scratch.
Then, I repeated the remaining steps. However, this time I used milk (1% in TBST for each antibody solution) instead of EveryBlot blocking buffer for each step. Milk is an incredibly effective blocking buffer as it’s packed with proteins, so hopefully it’s more effective at blocking background noise than EveryBlot was. I also left it in a new primary antibody solution I made (using milk) in the cold room overnight this time to allow for the antibodies to have more time to bind to the proteins.
The results were as follows:
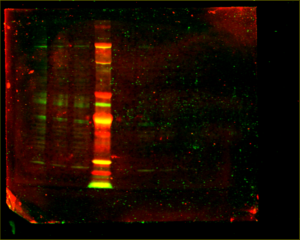
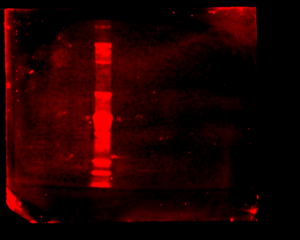
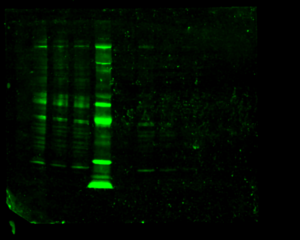
As the images still aren’t very clear, Steph speculates that the antibodies just aren’t very good and advised me to focus on data analysis and my project final product and presentation.
That’s it for this week! See you guys again next week for my (nearing final) blog!
Reader Interactions
Comments
Leave a Reply
You must be logged in to post a comment.
Hi! I have some more questions from another Honors Bio Student:
I wonder how long it takes to get results back from the mass spectrometry and how
they know which proteins are important.
How do you tell if the monomer and dimer forms of TMEM106B are doing something
different in the cells?
Another Honors Bio student asks:
How can you tell if the protein is alone or in a pair?
Why did you choose a mass spectrometry facility in Australia? Are there none in the US
that can do the same thing?
And another student:
-I wonder how disrupting dimer formation in TMEM106B impacts downstream
cellular processes or lysosomal function.
– How do you plan to differentiate between direct and indirect interaction partners
of TMEM106B in your IP experiments?
Last Honors Bio student:
What specific interaction partners did they find for the monomers versus the dimers?
Did they find anything surprising that could point to new treatments? I’m also curious
about how they worked with a lab in Australia for the mass spectrometry. How did they
send samples worldwide, and what was that collaboration like?
What was the biggest challenge you faced during this research, and was there a
moment when you felt like giving up or had to change your approach?